
Integrating Stereoelectronic Effects into Molecular Graphs: A Novel Approach for Enhanced Machine Learning Representations and Molecular Property Predictions



Traditional molecular representations, primarily focused on covalent bonds, have neglected crucial aspects like delocalization and non-covalent interactions. Existing machine learning models have utilized information-sparse representations, limiting their ability to capture molecular complexity. While computational chemistry has developed robust quantum-mechanical methods, their application in machine learning has been constrained by calculation challenges for complex systems. Graph-based representations have provided some topological information but lack quantum-chemical priors.
The increasing complexity of prediction tasks has highlighted the need for higher-fidelity representations. This work addresses these gaps by introducing stereo electronics-infused molecular graphs (SIMGs), which incorporate quantum-chemical interactions. SIMGs aim to enhance the interpretability and performance of machine learning models in molecular property predictions, overcoming the limitations of previous approaches and providing a more comprehensive understanding of molecular behavior.
Molecular representation is crucial for understanding chemical reactions and designing new materials. Traditional models use information-sparse representations, which are inadequate for complex tasks. This paper introduces stereoelectronics-infused molecular graphs (SIMGs), incorporating quantum-chemical information into molecular graphs. SIMGs enhance traditional representations by adding nodes for bond orbitals and lone pairs, addressing the neglect of essential interactions like delocalization and non-covalent forces. This approach aims to provide a more comprehensive understanding of molecular interactions, improving machine learning algorithms’ performance in predicting molecular properties and enabling evaluation of previously intractable systems, such as entire proteins.
The researchers employed Q-Chem 6.0.1 and NBO 7.0 for calculations using a high-throughput workflow infrastructure. They conducted Natural Bond Orbital analysis to quantify localized electron information, excluding Rydberg orbitals. The team introduced Stereo Electronics-Infused Molecular Graphs (SIMGs), incorporating stereoelectronic effects and representing donor-acceptor interactions. Their model architecture stacked multiple graph neural network blocks with graph attention layers and ReLU activation, addressing over-smoothing issues in multi-layer networks. Performance evaluation focused on lone pair classification and bond-related task predictions, demonstrating high accuracy and a 98% reconstruction rate of ground-truth extended graphs.
The model demonstrated exceptional performance across various prediction tasks, achieving high accuracy in classifying lone pair quantities and types. It successfully reconstructed the ground-truth extended graph in 98% of cases. Node-level tasks showed remarkable performance, with atom-related predictions achieving excellent R² scores and low MAEs and RMSEs. Lone pair predictions, especially for s and p-character, achieved excellent scores, while d-prediction tasks showed slightly lower performance due to limited data.
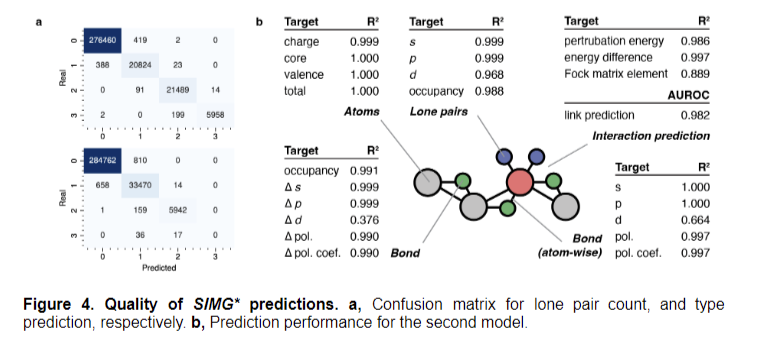
Bond-related task predictions were favorable, particularly for hybridization characters and polarizations. Performance positively correlated with interaction sample abundance. The F1 score ensured unbiased measurements for imbalanced classifications, highlighting the model’s effectiveness in capturing long-range interactions. These results underscore the successful integration of stereoelectronic effects into molecular graphs, significantly enhancing the model’s predictive capabilities across various molecular properties while also addressing challenges associated with d-character predictions.
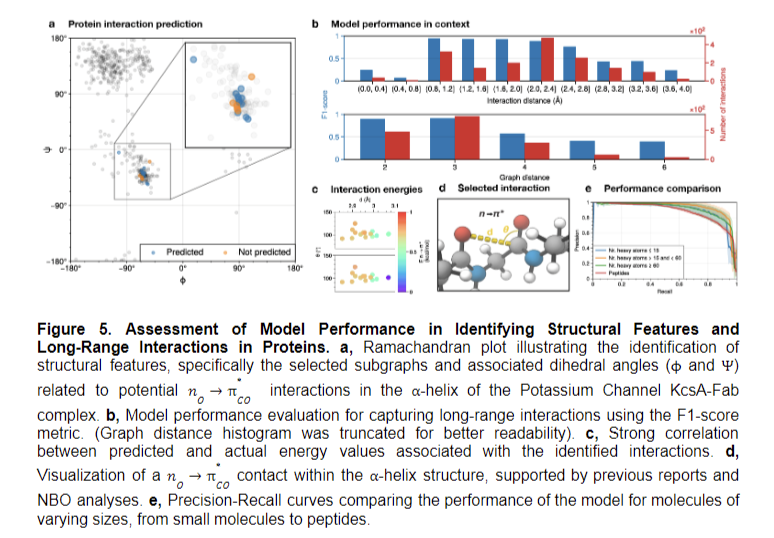
The study concludes that incorporating stereoelectronic interactions into molecular graphs significantly enhances machine-learning model performance, enabling a detailed understanding of molecular properties and behaviors. This approach allows predictions for previously inaccessible molecules, including complex biological structures. The new representation facilitates high-throughput Natural Bond Orbital analysis, potentially accelerating theoretical chemistry research. The tailored double-graph neural network workflow enables the broad application of learned representations. These findings suggest further exploration of stereoelectronic effects could lead to more sophisticated models, expanding applications in drug discovery and materials science. The study demonstrates the potential for advanced molecular representations to revolutionize predictive capabilities in chemistry and related fields.
Check out the Paper and GitHub. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. If you like our work, you will love our newsletter..
Don’t Forget to join our 48k+ ML SubReddit
Find Upcoming AI Webinars here

Shoaib Nazir is a consulting intern at MarktechPost and has completed his M.Tech dual degree from the Indian Institute of Technology (IIT), Kharagpur. With a strong passion for Data Science, he is particularly interested in the diverse applications of artificial intelligence across various domains. Shoaib is driven by a desire to explore the latest technological advancements and their practical implications in everyday life. His enthusiasm for innovation and real-world problem-solving fuels his continuous learning and contribution to the field of AI

Comments are closed, but trackbacks and pingbacks are open.